L Qin1,
S Xifa1,
X Dawei1,
X Yangjing1,
J Kangting1,
X Jian2,
Z Suqin1 ![]()
For correspondence:- Z Suqin Email: zhangsuqinwz@163.com Tel:+8657788002213
Received: 7 June 2016 Accepted: 17 October 2016 Published: 29 November 2016
Citation: Qin L, Xifa S, Dawei X, Yangjing X, Kangting J, Jian X, et al. Role of hypoxia-inducible factor in diabetic myocardial hypertrophy. Trop J Pharm Res 2016; 15(11):2421-2427 doi: 10.4314/tjpr.v15i11.17
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: This study was carried out to investigate the role of hypoxia-inducible factor (HIF) in diabetic cardiomyopathy in vitro.
Methods: Hypoxia was induced chemically in H9C2 cells (cardiac hypertrophy model), and the cells were treated with phenylephrine (PE), deferoxamine (DFO), PE + DFO, and HIF-1α siRNA under conditions of high and normal glucose. Western blot was used to analyze the ex
Results: PE caused hypertrophy in H9C2, which was ameliorated by HIF-1α. Compared to normal, under prolonged high glucose, the low ex
Conclusion: HIF-1 mediates diabetic myocardial hypertrophy, probably as a function of the degree of high glucose exposure and hypoxia.
Introduction
Cardiac hypertrophy is one of the characteristic features of heart failure [1,2]. In cardiac hypertrophy, growth of cardiac tissue and maintenance of its function are associated with capillary angiogenesis. The survival of endothelial cells is mainly maintained by growth factors including vascular endothelial growth factor (VEGF) [3,4], which plays an important role in the regulation of physiological and pathological processes in angiogenesis. It is known that specific knockout of VEGF can lead to decreased density of myocardial capillaries and systolic dysfunction [5,6]. In pathological cardiac hypertrophy, myocardial cells hold back the release of VEGF and bring down angiogenesis, thereby contributing to lack of oxygen and increased oxygen consumption. The hypoxia caused by this process firstly results in elevation of hypoxia inducible factor (HIF), which in turn leads to increases in levels of VEGF and other angiogenic factors. This adaptive response delays progression from pathological cardiac hypertrophy to heart failure. In early cardiac hypertrophy, stability of HIF-1 promotes glycolysis, which improves glucose utilization and allows for maintenance of normal functioning of the heart [7].
HIF is an important transcription factor involved in the regulation of about 300 genes which play important roles in the development of tissues and organs, proliferation and differentiation of cells, cellular energy metabolism, tumorigenesis and tumor progression [8,9]. HIF has three α subunits (HIF-1α, HIF-2α and HIF-3α) and a β subunit [10,11]. In general, the β subunits in stable expression, and is usually polymerized with HIF-α to form a heterodimer, which is transferred to the nucleus. HIF-1 is degraded through the ubiquitin-proteasome pathway, which process is mediated by the binding of HIF-1 and pVHL [12]. However, under hypoxic conditions, HIF-1 cannot be recognized by pVHL, a situation which helps HIF escape from degradation and get transported to the nucleus for binding to hypoxia response element (HRE) and the coactivator p300. Then the transcription of HIF-1 is initiated to offer the molecular foundation of the rapid response to hypoxia. It has been reported that HIF-1α expression in patients with ischemic heart disease is increased, whereas systemic knockout of HIF-1α can lead to cardiac hypertrophy and even death of embryos. Moreover, cardiac-specific knockout of HIF-1α can bring about changes in angiogenesis, energy availability, calcium flux and cardiac systolic function under normal oxygen conditions [13].
A large number of reports indicate that diabetes is closely related to angiogenesis. Diabetes or hyperglycemia disrupts HIF-mediated cardiac hypertrophy adaptive regulatory mechanism [14]. In diabetic retinopathy, abnormal increase of angiogenesis is directly related to elevated VEGF expression [15]. However, reduced expression of VEGF and its receptor in people with diabetic trauma, induces insufficient angiogenesis [16,17].
Cardiac hypertrophy caused by cardiac overload increases oxygen consumption, but when the amount of oxygen is reduced, it results in cardiac hypoxia. This process accelerates the activation of HIF-1, resulting in increase in levels of VEGF and other angiogenic factors and enhancement of glucose metabolism, which promotes angiogenesis and energy metabolism. These events delay the progression from cardiac hypertrophy to heart failure. So far, many studies have shown that diabetes is harmful to the heart. Short-term hyperglycemia may promote HIF-1 activation, which in turn increases the expression of VEGF, HXK-2, Glut-1 and enolase [18], but long-term hyperglycemia damages the function of HIF-1.
The role of HIF in diabetic cardiac hypertrophy is essentially unexplored. This study was carried out to investigate if HIF plays any role in diabetic cardiomyopathy in vitro.
Methods
Induction of cardiac hypertrophy
Cardiac hypertrophy was induced in H9C2 cardiomyocytes with phenylephrine (PE) (Sigma) [19], and deferoxamine (DFO) (Sigma) was used to induce the expression of HIF. HIF-1α was silenced with siRNA technology. The cells were cultured in normal-sugar medium and high-sugar medium (Invitrogen). Under three states of normal glucose (NG), high glucose (HG) and HIF1-α silence the expression of HIF-1α, HIF-2α, Glut-1, enolase, HXK-2, Bax and Bcl-2 in H9C2 cardiomyocytes were measured [20], as well as apoptosis of the H9C2 cells.
Cell line and culture
We used H9C2 cardiomyocytes from ATCC. The cells were cultured in DMEM containing 10 % heat-inactivated fetal bovine serum and 1 % penicillin–streptomycin mixture, and were grown at 37 °C under a humidified atmosphere of 5 % CO2 and 95 % air.
HIF-1α silencing
We dissolved 200 pM siRNA in a selective medium MEM, which was mixed with liposome 2000 for 25 minutes at room temperature. This was then added to each well of a six-well plate containing 5 × 105 H9C2 cardiomyocytes, and incubated for 4-6 h. Following that, antibiotic-free DMEM medium was replaced after 24 h incubation.
Western blot analysis
PE, PE + DFO, PE + DFO and HIF-1α siRNA were separately used to treat the H2C9 cardiomyocytes under conditions of high glucose (HG) and normal glucose (NG) for 48 h. The cells were harvested and lysed in RIPA buffer with protease inhibitors. Protein concentration was determined by the Coomassie brilliant blue method. Equal amounts of total cell lysates (60 mg protein) and Full-Range Rainbow Molecular Weight Marker were resolved using 12 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes through electrophoresis. PVDF membranes were immersed in blocking solution (5 % skim milk) to block non-specific binding for 1 h at room temperature. Thereafter, the PVDF membranes were immuno-blotted with primary monoclonal antibodies at 1:300 dilution at room temperature for 2 h, then overnight at 4 oC. Following this, further incubation was conducted for 2 h at room temperature with rabbit anti-mouse horseradish peroxide (HRP)-conjugated secondary antibody (1:3000 dilution). The protein-antibody complex was detected by enhanced chemiluminescence detection system.
Flow cytometry
After digesting with EDTA-free trypsin, the H9C2 cells were centrifuged at 1000 rpm for 5 min. After discarding the medium, the cells were washed three times with PBS. Thereafter an appropriate amount of binding buffer was added to achieve a dilution resulting in a concentration of 105 cells per 100 μL. In the dark, 5 μL Annexin V-FITC and 3 μL PI were added to 100 μL cells and stained for 10 min. Following that, 400 μL binding buffer was added. The fluorescence intensity of FITC was determined (λEx/Em = 488/515 nm), and apoptosis of cells was determined by fluorescence intensity of PI (λ = 560nm).
Statistical analysis
Experimental data are expressed as mean ± standard deviation (SD) after quantitation or semi-quantitation. Comparisons were analyzed using one-way analysis of variance (ANOVA). P < 0.05 was taken as statistically significant. Statistical analysis was performed using SPSS 17.0 while plots were made with GraphPad Prism5.
Results
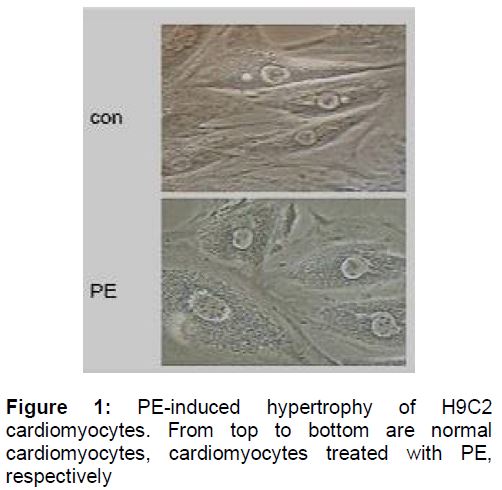
Phenylephrine-induced hypertrophy of H9C2
Under an optical microscope (), the control displayed normal features of H9C2 cells, with spindle shape, round and large-centered nucleus and small cell interval. H9C2 cardiomyocytes incubated with PE for 48 h were much larger and circular, round or elliptical in shape. Moreover, significant expansion of the cytoplasm, large cell interval and bigger nucleus were observed.
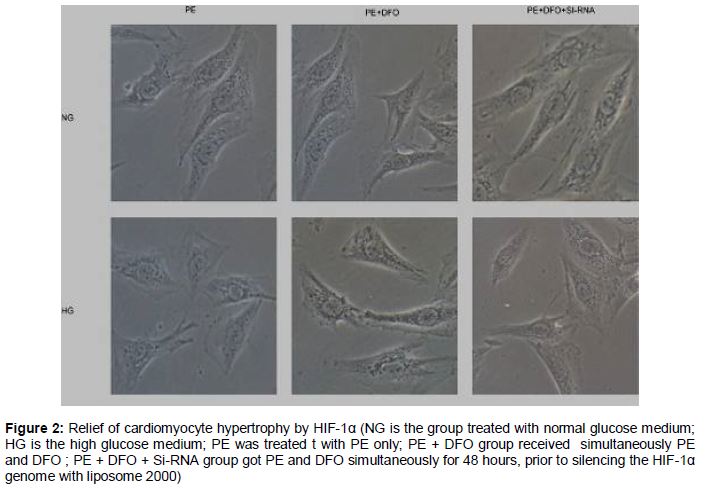
HIF-1α relieved cardiomyocytes hypertrophy
After culturing in 1 % serum medium for 12 h, H9C2 cells were treated with PE (100 μM) and DFO (200 μM) respectively for 48 h. Then HIF-1α gene was silenced with liposome 2000. Under an optical microscope (), in the case of NG, PE caused mild hypertrophy of myocardial cells, which was relieved after adding DFO to induced HIF-1α. In the HG group, cardiomyocytes treated with PE exhibited obvious myocardial hypertrophy, which was not mitigated by adding DFO due to HG-blockage of HIF-1α activation. Moreover, the hypertrophy in myocardial cells with silenced HIF-1α was almost unchanged in morphology.
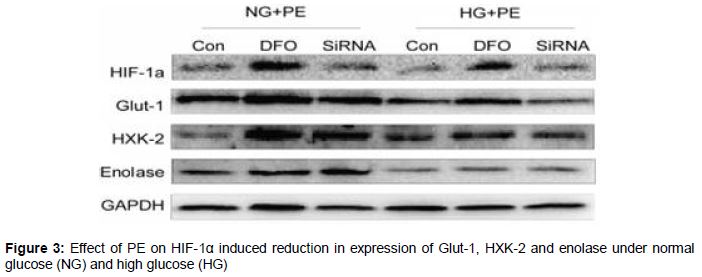
Effect of HIF-1α on expression of proteins associated with glycolysis
As shown in , silencing of HIF-1α gene resulted in reduced expression of Glut-1, HXK-2 and enolase. In the NG and HG groups, myocardial hypertrophy occurred after treatment with PE, and in the groups administered with DFO, HIF-1α expression was relatively higher than in the groups treated with PE only. In addition, SiRNA reduced the expression of HIF-1α. HG decreased the activation of HIF-1α, which led to lower expression of HIF-1α in this group than in the NG group. Administration of DFO to the HG group resulted in corresponding decrease in the expression of Glut-1. Long-term HG treatment led to decreased expression of HXK-2 but treatment with DFO increased the expressions of HXK-2 and enolase.
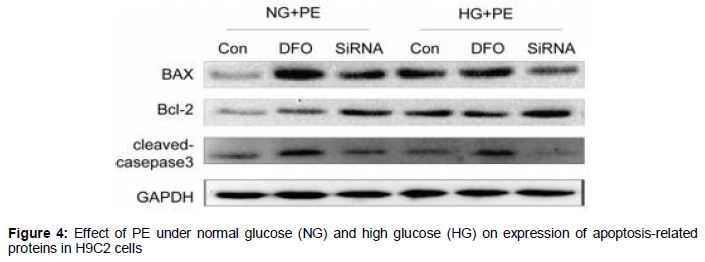
Effect of HIF-1α on the expression of proteins associated with apoptosis
As shown in , compared with the normal group, HIF expression was reduced, while Bax increased, Bcl-2 decreased and activated caspase3 was higher in the long-term HG group.
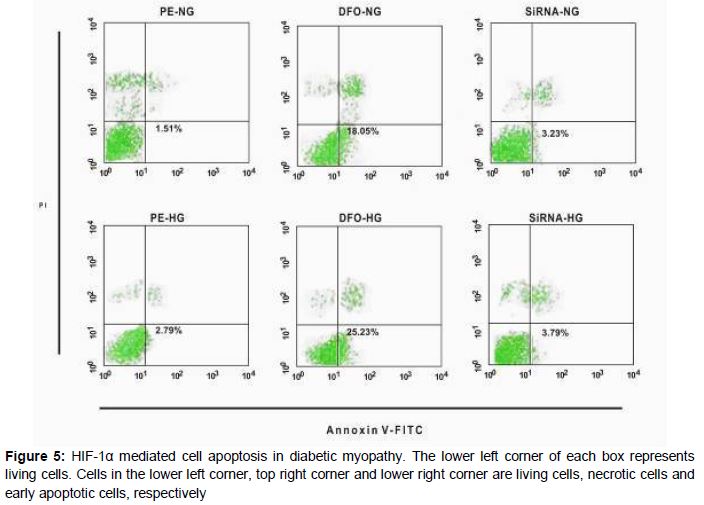
Evidence of apoptosis from flow cytometry
Results from flow cytometry are shown in . HIF-1α mediated the apoptosis of the hypertrophic cardiomyocytes exposed to short-term high glucose concentrations.
Discussion
Previous studies have shown that high expression of heart HIF-specific transgene prevented myocardium structural disorder and myocardial fibrosis in mice and exhibited the decrease of VEGF, HXK-2 and Glut-1 caused by diabetes [18]. Enhanced HIF-1α promotes antioxidant capacity of the heart and prevents the occurrence of diabetic cardiomyopathy. However, cardiac hypertrophy associated with pressure lowers HIF-1α [7]. Cell culture studies have demonstrated that high glucose conditions block HIF-1α activation, thereby blocking adaptation to hypoxia. The current study is based on earlier investigations on the role of HIF in myocardial hypertrophy under high glucose conditions in vitro cell culture experiments [19,21,22]. We used PE to establish the cardiac hypertrophy model, and DFO was given to mimic a hypoxic environment necessary for HIF production. Moreover, gene silencing technology was used to silence HIF-1α. Under NG, the PE-treated cells displayed slight hypertrophy, but after treatment with DFO, cell morphology was restored. While HG made myocardial hypertrophy more obvious, even DFO-induced HIF-1α did not significantly mitigate the hypertrophy. This is consistent with previous studies stating that high glucose is detrimental to the activity of HIF [18]. HIF-1α silencing did not change cell morphology substantially, which further indicates that HIF-1α has a protective effect on myocardial hypertrophy [18].
In the pathogenesis of heart failure, as well as the physiological function of blood vessels, significantly altered glucose and lipid metabolism is associated with heart failure [14]. The transition from aerobic metabolism to glycolysis is one of the key cellular processes for adaption to hypoxia response, which is regulated by HIF-1. Therefore, we examined the expression of glycolytic proteins using Western Blot. The decrease in HIF-1α resulted in reduced expression of Glut-1, HXK-2 and enolase. Hypoxia can easily induce apoptosis, so it was necessary to study the expressions of apoptosis-related proteins as well. The results obtained showed that Bax expression was increased, while that of Bcl-2 decreased, and activated caspase 3 was higher under long-term high glucose condition. The results suggest that high glucose levels impair HIF activity, and that HIF protected the H9C2 cells from apoptosis.
However, results of flow cytometry indicated that HIF-1 promoted cell apoptosis under short-term high glucose condition, which is at variance with the western blot results on apoptosis-associated proteins. The reason for this apparent contradiction is not very clear, but it may be related to degree of high glucose exposure and hypoxia.
Conclusion
Short-term hyperglycemia may contribute to activation of HIF-1, which in turn increases the expression of VEGF, HXK-2 and Glut-1. However, long-term high blood sugar may be detrimental to the function of HIF-1. It can be inferred from our results that HIF-1 is likely to play an important role in diabetic cardiac hypertrophy. On account of different effects of HIF-1 under different glucose loads and hypoxia, it is concluded that HIF may play a pathogenic role in heart failure.
Declarations
Acknowledgement
References
Archives


News Updates